| Name | Last modified | Size | Description | |
|---|---|---|---|---|
| Parent Directory | - | |||
| config.toml | 2023-09-28 15:07 | 549 | ||
| Mo2S4.py | 2023-09-28 15:08 | 669 | ||
| ground-state.toml | 2023-09-28 15:09 | 780 | ||
| Mo2S4.json | 2023-09-28 15:08 | 827 | ||
| Mo2S4.cif | 2023-09-28 15:08 | 875 | ||
| settings.toml | 2023-09-28 15:11 | 1.3K | ||
| material_modeling-log.txt | 2023-09-28 15:11 | 1.3K | ||
| MoS2_OCD_9007660.cif | 2023-09-28 15:07 | 1.4K | ||
| Mo2S4.html | 2023-09-28 15:08 | 2.0K | ||
| Mo2S4.png | 2023-09-28 15:08 | 5.5K | ||
| README.html | 2023-09-28 15:11 | 6.2K | ||
| properties.toml | 2023-09-28 15:11 | 12K | ||
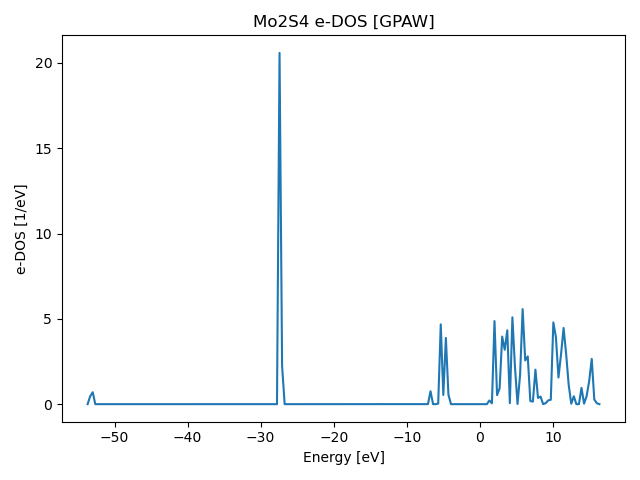
| electronic_dos.svg | 2023-09-28 15:09 | 21K | ||
| bravais_lattice.svg | 2023-09-28 15:08 | 23K | ||
| calculator.log | 2023-09-28 15:11 | 23K | ||
| electronic_dos.png | 2023-09-28 15:09 | 25K | ||
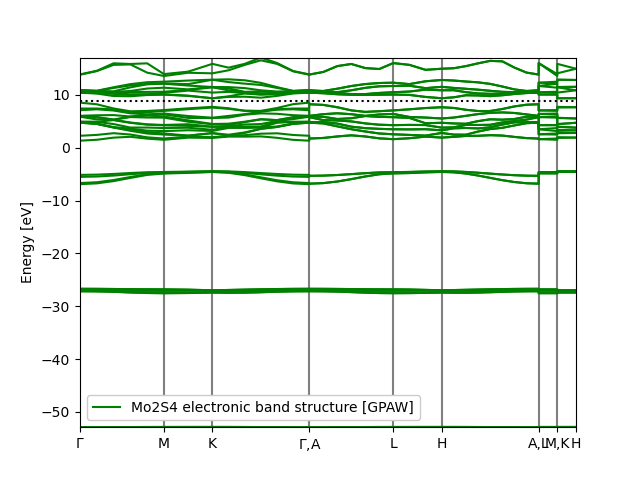
| electronic_band_structure.json | 2023-09-28 15:11 | 27K | ||
| properties.h5 | 2023-09-28 15:11 | 41K | ||
| electronic_band_structure.png | 2023-09-28 15:11 | 54K | ||
| electronic_band_structure.svg | 2023-09-28 15:11 | 63K | ||
| calculator.gpw | 2023-09-28 15:09 | 1.5M | ||
This page presents an overview of the calculation performed for the material Mo2S4.
The following computations are requested:
The calculator being used is: GPAW which detailed configuration as follows:
{'parallel': {'kpt': None, 'domain': 1, 'band': 1, 'order': 'kdb', 'stridebands': False, 'augment_grids': False, 'sl_auto': False, 'sl_default': None, 'sl_diagonalize': None, 'sl_inverse_cholesky': None, 'sl_lcao': None, 'sl_lrtddft': None, 'use_elpa': False, 'elpasolver': '2stage', 'buffer_size': None}, 'timer': , 'scf': None, 'wfs': None, 'density': None, 'hamiltonian': None, 'spos_ac': None, 'observers': [], 'initialized': False, 'world': , 'log': , 'reader': None, 'atoms': None, 'results': {}, 'parameters': {'mode': 'pw', 'xc': 'PBE', 'occupations': {'name': 'fermi-dirac', 'width': 0.13}, 'poissonsolver': None, 'h': None, 'gpts': None, 'kpts': (3, 3, 3), 'nbands': None, 'charge': 0, 'setups': {}, 'basis': {}, 'spinpol': None, 'filter': None, 'mixer': None, 'eigensolver': 'rmm-diis', 'background_charge': None, 'experimental': {'reuse_wfs_method': 'paw', 'niter_fixdensity': 0, 'magmoms': None, 'soc': None, 'kpt_refine': None}, 'external': None, 'random': False, 'hund': False, 'maxiter': 333, 'symmetry': 'off', 'convergence': {'energy': 1e-05}, 'verbose': 0, 'fixdensity': False, 'dtype': None}, '_directory': '.', 'prefix': None, 'name': 'gpaw'} The setting used are:
{'atoms': '/var/www/html/data/material_modeling/MoS2_OCD_9007660.cif-GPAW-paveldudin-20230928-150759/MoS2_OCD_9007660.cif', 'directory': '/var/www/html/data/material_modeling/MoS2_OCD_9007660.cif-GPAW-paveldudin-20230928-150759/', 'mpi': 8, 'start_date': 'Thu Sep 28 15:08:02 2023', 'compute': 'ELECTRONS', 'log': None, 'calculator': 'GPAW', 'convergence': 1e-05, 'displacement': 0.01, 'ecut': None, 'eigensolver': 'rmm-diis', 'maxiter': None, 'nbands': None, 'occupations': 0.13, 'potentials': None, 'xc': 'PBE', 'smearing': 'semiconductor', 'user_id': 'paveldudin', 'user_ip': ' [192.168.3.58]', 'mpirun': '/usr/bin/mpirun -np 8', 'chemical_symbols': ['Mo', 'Mo', 'S', 'S', 'S', 'S'], 'chemical_formula': 'Mo2S4', 'supercell': array([2, 2, 2]), 'kpoints': array([3, 3, 3]), 'kpts_density': 1296, 'label': 'Mo2S4', 'Atoms': Atoms(symbols='Mo2S4', pbc=True, cell=[[3.161, 0.0, 0.0], [-1.5804999999999993, 2.7375063013626106, 0.0], [0.0, 0.0, 12.295]], spacegroup_kinds=..., calculator=GPAW(...))}
Properties:
{'magnetic_moment': 0.0, 'kinetic_energy': 0.0, 'potential_energy': -43.78505761517259, 'stress': array([-3.18665996e-02, -3.18633065e-02, -6.40308423e-02, -8.07755017e-06,
4.66045286e-06, -2.29280271e-06]), 'momenta': array([[0., 0., 0.],
[0., 0., 0.],
[0., 0., 0.],
[0., 0., 0.],
[0., 0., 0.],
[0., 0., 0.]]), 'total_energy': -43.78505761517259, 'angular_momentum': array([0., 0., 0.]), 'charges': None, 'dipole_moment': array([-8.39663339e-05, 1.45394819e-04, 9.38522171e-08]), 'moments_of_inertia': array([ 256.14930144, 3325.99042454, 3582.13972595]), 'center_of_mass': array([0.79024526, 1.36876136, 6.1475 ]), 'Fermi_level': 8.684060853129363}

The computation results are available in an HDF5 file. View it with silx view properties.h5
{kind=link}
{kind=link}
{kind=link}